Shark Cartilage
Shark cartilage
Journal of Clinical Oncology, Vol16, No 11(November), 1998
Phase I/II Trial of the Safety and Efficacy of Shark Cartilage in the Treatment of Advanced Cancer.
Denis R. Miller, Gary T. Anderson, James J. Stark, Joel L. Granick, and DeJuran Richardson
PurposePatients with cancer and chronic inflammatory disorders have used shark cartilage (SC) preparations for many years. Preclinical studies that support their beneficial effects are scanty, and reports of clinical trials have been anecdotal. The proposed mechanisms of antitumor action include direct or indirect inhibition of angiogenesis. Because of the emerging use of SC as an alternative to conventional cancer therapy, this trial was launched to evaluate the safety and efficacy of SC. Patients and Methods
Sixty adult patients with advanced previously treated cancer (breast, 16 patients; colorectal, 16 patients; lung, 14 patients; prostate, eight patients; non-Hodgkin lymphoma, three patients; brain, one patients; and unknown primary tumor, two patients) of diagnosis, resistance to conventional therapy, objective measurable disease, life expectancy of 12 weeks or greater, Eastern Cooperative Oncology Group (ECOG) anticancer therapy, no prior SC, and informed consent. Patients underwent evaluation of the extent of disease, quality-of-life score (Functional Assessment of Cancer Therapy-General [EACT-G] scale), and hematologic, biochemical, and selected immune function studies at baseline and after 6 and 12 weeks of SC therapy. The dose of SC was 1 g/kg daily orally in three divided doses. Standard criteria were used to evaluate adverse events and response. Results
Ten of 60 patients were lost to follow-up(LTFU) or refused further treatment(RFT) before the 6-week evaluation and were not assessable for toxicity and response. Three patients with stable disease at 6 weeks were LTFU or RFT thereafter. Of the 47 fully assessable patients, five were taken off study because of gastrointestinal toxicity or intolerance to SC. Progressive disease (PD) at 6 or 12 weeks occurred in 22 and five patients, respectively. Five patients died of PD while undergoing SC therapy. No complete (CRs) or partial responses (PRs) were noted. Median time to tumor progression in the entire study population was 7 +- 9.7 weeks (mean, 11.4 weeks; range, 3.7 to 45.7 weeks.) Ten(20%) of 50 assessable patients, or 16.7% of the 60 intent-to treat patients, ahd stable disease (SD) for 12 weeks or more. The median time to tumor progression was 27 weeks, the mean was 28.8 +- 9.9 weeks, and the range was 18.6 to 45.7 weeks. In this subset, FACT-G scores improved in four patients, were unchanged in four patients, and declined in two patients. Twenty-one adverse events (grade 1, egith events; grade 2, seven events; and grade 3, six events) were recorded, 14 of which were gastroenterologic (nausea, vomiting, constipation). Conclusion
Under the specific conditions of this study, SC as a single agent was inactive in patients with advanced-stage cancer and had no salutary effect on quality of life. The 16.7% rate of SD was similar to results in patients with advanced cancer treated with supportive care alone. 1998 by American Society of Clinical Oncology.
- metabolic, pharmaceutical, herbal, or immune therapies
- dietary alterations or supplementation with high-dose multivitamins, minerals, antioxidants, or other nonnutrient agents
- alternative therapists or healing methods (eg, acupuncture, hypnosis, spiritual therapy, mind-body therapy)
Eligibility Criteria
Adults patients (aged >=15 years) with histologically confirmed, advanced-stage (stage III to IV), previously treated, refractory, recurrent, and/or metastatic cancer were entered onto the study from January 1995 through September 1996. Areview of submitted sliders, surgical pathology reports, or surgical specimens or needle biopsies of fresh tissue confirmed the diagnosis of cancer. Only patients with stages III to IV cancers of the lung, breast, colorectum, prostate, bladder, and brain and non-Hodgkin's lymphoma were eligible. The interval between prior anticancer therapy and entry onto the study was 3 weeks. Patients with primary brain tumors required an interval of 45 days from prior cranial radiation therapy before enrollment. The protocol was amended in September 1995 to permit the entry of patients who refused conventional anticancer therapy after being fully informed of the available conventional therapies and their greater likelihood of efficacy. All patients had objective measurable disease determined by physical examination and appropriate medical imaging studies. Patients with only increased levels of a surrogate biologic marker of persistent or recurrent disease (eg. Elevated prostate-specific antigen levels without measurable disease) were ineligible. All patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2 and a projected life expectancy of at least 12 weeks. SC was the only taken or were taking SC at 20% or greater of the protocol prescribed dose (1 g/kg daily) for 1 month or more were excluded. Pregnant and lactating women were also excluded. The protocol was approved by the and the Institutional Review Boards of the two participating sites (Midwestern Regional Medical Center, Zion, IL, and Cancer Treatment Centers of America at Maryview Hospital, Prtsmouth, VA). Informed consent was obtained from all patients enrolled onto the study. The SC used in the study (Cartilade) was generously provided to study patients by Cartilage USA, Elmsford, NY.Study Design
Baseline evaluation included a complete history and physical examination. The specific details of all prior anticancer therapy were obtained, which included modality of treatment, first and last date of treatment, surgical procedures, dose schedule of all chmotherapeutic agents, total dose, and response. Baseline laboratory and imagin studies included complete blood count and differential count, chemistry profile (SMA-21), prothrombin time, partial thromoboplastin time, and immune function panel (T and B cell subsets, natural-killer cell function, and serum immunoglobulin, tumor nectosis factor, and interleukin-2 levels). Tumor markers and medical imaging studies appropriate for the specific tumor site were obtained to document the extent of disease. Cross-dimensional diameters of all measurable lesions were determine whenever possible. Assessable but nonmeasurable lesions (eg, positive bone scans) were monitored. Quality of life was quantified with the previously validated Functional Assessment of Cancer Therapy-General (FACT-G) scale. This self-administered questionnaire evaluates social, physical, social/family, emotional, and functional well-being and professional relationships and is sensitive to clinical change. Patients were treated with SC powder, 1g/kg daily orally in three devided doses taken before meals. The powder (5 g/tsp) was mixed in fruit juice. Vanilla flavor (2% w/w of SC pwder) was added by the manufacturer approximately 6 months after the study was opened, but no adjustment in dosage was made.Shark cartilage in advanced cancer
Patients were treated as outpatients and returned after 6 and 12 weeks for interval history, physical examination, laboratory and imaging studies, FACT-G score, evaluation of response, and documentation of adverse events. Patients received a 6-week supply of SC after the initial baseline evaluation. Compliance was determined by direct questioning of patients and confirmation of pharmacy records. The dose of SC was increased of 1.3 g/kg daily (30%) if there was no measurable response after 6 weeks of therapy. All patients received comprehensive meicaloncology care commensurate with their needs. This included active psychosocial support and a standard nutritional supplementation program that consisted of multivitamins, minerals, and antioxidants. Trained registered dietitians provided the specific details of a low-fat, high-vegetable, and high-fruit diet. Standard criteria were used to evaluate objective clinical activity, subjective responses (quality of life), and toxicity (National Cancer Institute criteria). A complete response (CR) was defined as the complete disappearance of all tumor messes without the appearance of any new lesions and normalization of all clinical and laboratory signs and symptoms of active disease. A partial response (PR) was defined as a 50% or greater reduction of the products of the longest perpendicular diameters of the measured sentinel lesions without demonstrable new lesions elsewhere. Stable disease (SD) occurred when no new lesions appeared and no measurable lesions increased more than 25% in a cross-directional area. Progressive disease (PD) was defined as the appearance of new lesions and /or increase in the cross-sectional area of any previously known lesion by greater than 25%. An adequate trial was 12 weeks of therapy. Off-study criteria were (1) pd, (2) the patient's desire to withdraw, (3) noncompliance, (4) unusual or unacceptable toxicity, or (5) emerging evidence that SC was of no benefit to patients with a similar tumor type. The end points of the evaluation included response rate, time to tumor progression (date on study to documented PD), and quantitative changes in FACT-G scores.Statistical Analysis
The trail was designed to enroll 12 assessable patients for each of seven tumor types, with an initial entry of 84 patients. Further accrual would be halted if no CR or PR were observed in each specific tumor type and the therapy would be judged inactive. If one or more CRs or PRs occurred among the 12 patients, another 25 patients were to be entered. Tumor types with four or more CRs or PRs were to be targeted for further study. The planned accrual was designed to provide a 90% likelihood of rejecting treatment with a true response rate of 5% or less of 20% or more. Data compiled at baseline, 6 weeks, and 12 weeks were analyzed by two-way analysis of variance by ranks. Preenty data of patients who became lost to follow-up (LTFU), withdrew, or refused further treatment were analyzed to determine if any features distinguished them from the other patients. Median values of quantities, such as age and days since diagnosis, were compared using Wilcoxon's rank-sum test, and categoric values (of quantities such as primary tumor site and off-study reason) were compared using ×2 tests and, for 2 × 2 tables, Fisher's exact tests.Results
Response
The characteristics to the 60 study patients are listed in Table 1. There were 24 men (40%) and 36 women (60%). The median age of all patients was 63 years. Ninety-seven percent of the patients had stage IV disease. The two patients with unknown primary tumors at the time of study entry had probable stage IV breast cancer. The mean ECOG performance score was 1.02. One patient with non-small-cell lung cancer had not prior anticancer therapy.| Tumor | |||||||
| Brain | Breast | Colon | Lung | Lymph | Prostate | Total | |
| Total no. entered | 1 | 18 | 16 | 14 | 3 | 8 | 60 |
| Assessable | 1 | 13 | 10 | 14 | 2 | 8 | 47 |
| Men | 1 | - | 6 | 8 | 1 | 8 | 24 |
| Women | - | 18 | 10 | 6 | 2 | - | 36 |
| Stage3 | 1 | - | - | 1 | - | - | 2 |
| Stage4 | - | 18 | 16 | 13 | 3 | 8 | 58 |
Note: Stage not specified in 6 patients.
Ten patients were not assessable for response or toxicity because they were LTFU or refused further
therapy (RFT) before 6 weeks of treatment. Their median age (54 years) was significantly younger than
the median age (63 years) of the overall study population.
Three patients with SD at 6 weeks were LTFU between 6 and 12 weeks. Patients who were LTFU, withdrew,
or RFT were mostly women (10 v three). Expect for age, there were no distinguishing features between
these groups and the entire study population.
The response and disposition of patients by tumor type are listed in Table 2. Five patients withdrew
because of gastrointestinal toxicity. Five patients died of PD while undergoing therapy. Twenty-two
patients (36.7%) had PD before or at the 6-week evaluation point (range, 4 days to 6 weeks). Five
patients developed PD between 6 and 12 weeks of therapy. No CRs or PRs were observed, but 10 to 50
assessable patients (20%) had SD for 12 weeks or more (range, 12 to 45.7 weeks). The time to tumor
progression of all study patients is shown in Fig 1.
| Response/ Off Study | Brain | Breast | Colon | Lung | Lymphoma | Prostate | Total |
| PD>=12Weeks(SD) | - | 2 | 3 | 3 | - | 2 | 10 |
| PD<12Weeks | - | 2+ | - | 2 | - | 1 | 5 |
| PD<=12Weeks(SD) | 1 | 6 | 5 | 5 | - | 5 | 22 |
| SD at 6 weeks | - | - | 1 | - | 2 | - | 3 |
| LFTU or RFT | - | 5 | 5 | - | - | - | 10 |
| Toxicity | - | 2 | 1 | 1 | 1 | - | 5 |
| Death | - | 1+ | 1 | 3 | - | - | 5 |
| Total | 1 | 18 | 16 | 14 | 3 | 8 | 60 |
* Includes 2 patients with unknown primary tumors at entry but with metastatic breast cancer
on further evaluation. +Includes 1 patient with unknown primary tumor intially.
The characteristics of the 10 patients with SD for 12 weeks or more are listed in Table.3.
All had stage IV disease (colorectal, three patients; lung, three patients; breast, two patients;
prostate, two patients). The median time to tumor progression was 27 weeks, the mean +-SD was 28.8
+- 9.9 weeks, and the range was 18.6 to 45.7 weeks. Respective times for three patients were unavailable.
The prostate-specific antigen levels in the two prostate cancer patients did not decrease during SD.
One patients refused conventional therapy for non-small-cell lung cancer, started SC therapy 25.6 weeks
after diagnosis, and had PD after 25 weeks of treatment, during which time his FACT-G scores declined
from 84 to 67. One patient refused further therapy after 12 weeks.

Quality of Life
The FACT-G scores of the 25 assessable patients with complete data are shown in Fig 2. There was no significant difference between those patients with FACT-G scores and those without regarding preentry characteristics or response to treatment. Among those with complete data, the median change from the baseline score to the 6-week score was -1.3 and -4.2 to the 12-week score was -2.4 points. In general, no overall improvement in quality of life was observed. In the group of patients with SD, FACT-G scores improved in four patients, were unchanged in four patients, and declined in two patients. There was no relationship between time to tumor progression and FACT-G scores, but there was evidence of a trend between time since diagnosis and improvement in quality of life scores. Within this group, patients who entered the study within 65 weeks of diagnosis had median FACT-G scores at baseline (n=17), 6 weeks (n=8), and 12 weeks (n=4) of 79.3, 79.8, and 64.5, respectively. The corresponding median scores in the entire study population for which data were available were 84, 79, and 76, respectively. The small number precluded meaningful statistical analysis.Toxicity/Adverse Events
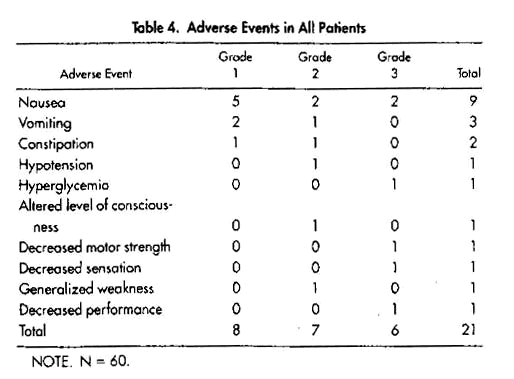
SD therapy with the dose schedule used in this study was generally well tolerated and was not associated with hematologic or biochemical toxicity. Of note, hypercalcemia was not observed. No patient developed hepatitis or abnormal liver function test.30 No effects were noted on measured immune function parameters (data not shown). Adverse events are recorded in Table 4. Toxicity was generally mild (grade 1) to moderate. Five episodes of grade 3 toxicity were recorded. Fourteen of the 20 reported adverse events were gastrointestinal (nausea, nine patients; vomiting three patients; constipation, two patients).